
Workflow Management - Pegasus
Objectives
- Learn how to use pegasus to complete a set of computational tasks.
Overview
Pegasus can handle millions of computational tasks and takes care of managing input/output files for you. In this section, we will learn how to run large scale NAMD simulations with pegasus. First, we will focus on a toy example of running single NAMD job. Then we extend our workflow to run N sequential jobs. Finally, we will see an example of running M parallel jobs where each parallel-job contains N sequential jobs.
Introduction
Pegasus enables scientists to construct workflows in abstract terms without worrying about the details of the underlying execution environment or the particulars of the low-level specifications required by the middleware. Some of the advantages of using Pegasus includes:
-
Performance - The Pegasus mapper can reorder, group, and prioritize tasks in order to increase the overall workflow performance.
-
Scalability - Pegasus can easily scale both the size of the workflow, and the resources that the workflow is distributed over. Pegasus runs workflows ranging from just a few computational tasks up to 1 million. The number of resources involved in executing a workflow can scale as needed without any impediments to performance.
-
Portability / Reuse - User created workflows can easily be run in different environments without alteration. Pegasus currently runs workflows on top of Condor, Grid infrastrucutures such as Open Science Grid and TeraGrid, Amazon EC2, Nimbus, and many campus clusters. The same workflow can run on a single system or across a heterogeneous set of resources.
-
Data Management - Pegasus handles replica selection, data transfers and output registrations in data catalogs. These tasks are added to a workflow as auxiliary jobs by the Pegasus planner.
-
Reliability - Jobs and data transfers are automatically retried in case of failures. Debugging tools such as pegasus-analyzer helps the user to debug the workflow in case of non-recoverable failures.
-
Provenance - By default, all jobs in Pegasus are launched via the kickstart process that captures runtime provenance of the job and helps in debugging. The provenance data is collected in a database, and the data can be summaries with tools such as pegasus-statistics, pegasus-plots, or directly with SQL queries.
-
Error Recovery - When errors occur, Pegasus tries to recover when possible by retrying tasks, by retrying the entire workflow, by providing workflow-level checkpointing, by re-mapping portions of the workflow, by trying alternative data sources for staging data, and, when all else fails, by providing a rescue workflow containing a description of only the work that remains to be done.
Single NAMD Job
The necessary files are available to the user by invoking the tutorial command.
$ tutorial ### Without an argument, it shows the list of available tutorials.
$ tutorial pegasus-namd ### The files to run the pegasus-namd tutorial are created under the directory tutorial-pegasus-namd
$ cd tutorial-pegasus-namd/Single ### First, we will focus on running single NAMD job with pegasus workflow management
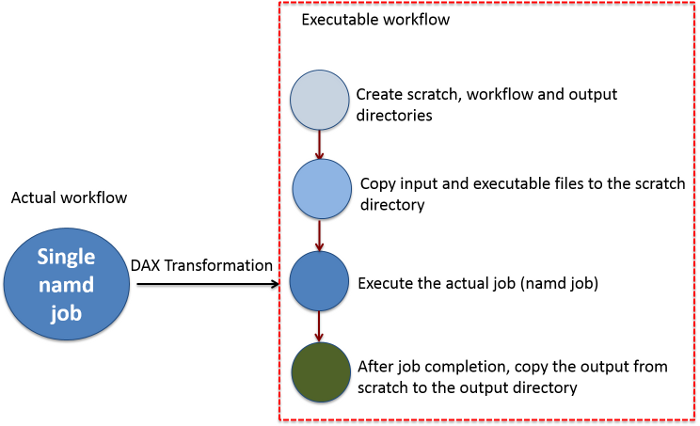
Figure 1. The actual workflow of executing single job is transformed into a set of jobs in pegasus workflow. Such a transformation is useful to keep track of input and output data. In particular, when we have to deal with lot of jobs.
Now we will take a closer look at the files to understand how to create the pegasus workflow for single job.
Input and output files
There are several files and directories under the path "tutorial-pegasus-namd/Single". Some files are required to run NAMD simulations and other files are related to pegasus workflow management. The files required by NAMD are under the directories
inputs/ ### Directory contains the input configuration file for NAMD
ExeFiles/ ### Directory contains Bash script file that executes the NAMD with relevant arguments
paramdirs/ ### Directory contains the structure, psf and topology files
The following files are related to pegasus workflow management
pegasusrc ### The configuration file for pegasus
dax-generator-singleJob.py ### Python script generates dax.xml file
sites-generator.bash ### Bash script generates sites.xml file
submit.bash ### Bash script submits the pegasus workflow.
The file "pegasusrc" contains the pegasus configuration information. We can simply keep this file in the current working directory without worrying much about the details ( If you would like to know the details, please visit the pegasus home page). The files - dax.xml and sites.xml contain the information about the work flow and data management.
Submit script
Let us pay attention to few parts of the "submit" script to understand about submitting the workflow. Open the file "submit" and take a look
...
line 9 ./dax-generator-singleJob.py ### Execution of "dax-generator-singleJob.py" script. Generates dax.xml.
...
line 14 ./sites-generator.bash ### Execution of "sites-generator.bash" script. Generates sites.xml.
...
line 17 pegasus-plan \ ### Executes the pegasus-plan with the following arguments
line 18 --conf pegasusrc \ ### pegasus configuration file
line 19 --sites condorpool \ ### jobs are executed in condorpool
line 20 --dir $PWD/workflows \ ### The path of the workflow directory
line 21 --output-site local \ ### Outputs are directed to the local site.
line 22 --dax dax.xml \ ### Name of the dax file
line 23 --submit ### Type of action is submit
sites-generator
The purpose of sites-generator script is to generate the sites.xml file. There are several lines declared in the sites-generator script. We need to understand the lines defining the scratch and output directories.
...
line 4 cat >sites.xml <<EOF ### creates the file "sites.xml" and appends the following lines.
...
line 11 <file-server protocol="file" url="file://" mount-point="$PWD/scratch"/> ### Define the path of scratch directory
line 12 <internal-mount-point mount-point="$PWD/scratch"/> ### Define the path of scratch directory
...
line 17 <file-server protocol="file" url="file://" mount-point="$PWD/outputs"/> ### Define the path of output directory
line 18 <internal-mount-point mount-point="$PWD/outputs"/> ### Define the path of output directory
...
line 32 EOF ### End of sites.xml file
The files "submit.bash" and "sites-generator.bash" will not change very much for a new workflow. We need to edit these two files, when we change the name of the dax-generator and/or the path of outputs, scratch and workflows.
DAX generator
The file - dax.xml contains the workflow information, including the description about the jobs and required input files. We could manually write the dax.xml file but it is not very pleasant for the human eye to deal with the xml format. Here, dax.xml is generated via the python script "dax-generator-singleJob.py". Take a look at the python script, it is self explanatory with lots of comments. If you have difficulty to understand the script, please feel free to send us an email. Here is the brief description about dax-generator python script.
...
line 8 dax = ADAG("namd-singleJob") ### Name of dax. You can change any interesting name you like.
...
line 11
... ### Defines the directory paths of base, exe, inputs and param directories
line 14
...
line 18
... ### Add the executable "namd_exe.bash" and its path to the dax.xml file
line 21
...
line 25
... ### Loops over the files in the paramdirs. Add the param files and their path to the dax.xml file
line 28
...
line 31
... ### These lines describe the namd conig file "ubq_gbis_eq.conf" and its location. The information about config file is included in dax.xml file in way
similar to param files were included.
line 35
...
line 38 ### A job "namdEq_job" is added in dax.xml file.
... ### Define the job, input and output files. Pegasus would transfer the INPUT files to the remote worker nodes before job execution. Pegasus would transfer
the OUTPUT files from the remote worker nodes after job execution.
line 47
...
line 50
... ### Although the path of the param files are defined in the lines 25-28, so far they are not defined as input files for the executable. Here, the param
files are defined as input files so that pegasus transfer these files to the remote machine.
line 53
Job submission and status
To submit the job
### To submit the job
$ ./submit.bash
To check the status of the submitted job
$ pegasus_status
### or you can also check with the condor_q command
$ condor_q username ### username is your login ID
Pegasus creates the following directories
scratch/ ### Contains all the files (including input, parameter and execution) required to run the job are copied in this directory.
workflows/ ### Contains the workflow files including DAGMan, data transfer scripts and condor job files.
outputs/ ### Where the NAMD output files are stored at the end of each job.
The path of the scratch, workflows and outputs directories are declared in the "submit" scripts at lines 19, 20, 25,26 and 47.
Exercise -1
Under the directory "tutorial-pegasus-namd/Exercises/SingleEx1" you will see relevant files to run the single NAMD job with pegasus. However, you need to change few things to run submit the job. The errors are associated with the definition of names of dax-generator and NAMD input files. You have to correct these two file names in the submit submit.bash and in the dax-generator script.
Exercise-2
Under the directory "tutorial-pegasus-namd/Exercises/SingleEx2" you will see relevant files. In this exercise, you have to specify the correct path for the scratch, output and workflow directories. All these information are included in the site-generator.bash and submit.bash
N-Sequential jobs
The current workflow of N sequential jobs demonstrates the ability to complete large scale molecular dynamics simulations with pegasus. We break the long-time scale simulation into several short-time scale simulations. The short simulations are performed in a sequence and then the results are combined to achieve the the long-time scale simulation. In this example, we will learn how to run N sequential NAMD jobs.
Here, the NAMD jobs are executed one-by-one. In these sequential executions, the restart files are utilized. A NAMD job generates the restart files that are necessary to start the next job. To run N-sequential NAMD jobs, we need N input files. We have to specify in the input file that the restart files from each simulation are available to the next job. So our first step is to generate N input files that are suitable to run N-sequential jobs.
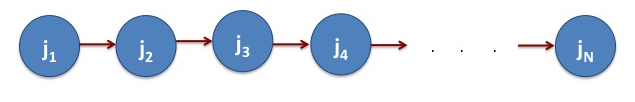
Figure 2. The workflow to run linear sequence of jobs. The blue circles represent jobs and the arrows represent direction of data flow. This means data from J1 is required to start the job j2, the data from j2 is required to start the job j3 and so on.
Generating N-sequential input files
We use a script to do the task of generating N input files to run the sequential molecular dynamics simulations.
$ cd tutorial-pegasus-namd/N-Sequential ### Directory of required files to run N-sequential MD jobs
$ cd InputGen ### the input template and input generation script are available in this directory
$ ./namd_gen_pegasus_input.bash 10 ### This would generate 10 configuration files for NAMD under the directory tutorial-pegasus-namd/N-Sequential/inputs
Basically, the script generates input files from a reference template. In these input files, the name of the restart files are specified in correct order that makes the input files suitable for sequential execution of NAMD jobs. DAX-generator We have to change our dax.xml via the dax-generator script to account that there are N-sequential jobs. Let us see where is our dax generator script.
$ cd tutorial-pegasus-namd/N-Sequential ### Main directory
$ ls dax*.py
dax-generator-namdEq-sequential.py ### This is the dax generator file.
Let us analyze the dax-generator script to find what are the primary differences in running N-sequential jobs Vs running single job. Open the file dax-generator-namdEq-sequential.py.
...
line 33 for i in range (0,10): ### We loop over the number of input files. Note that if there 20 jobs the range is 20 in the loop.
...
line 66 if job_count > 0: ### We check the number of jobs to define the job dependencies. For each input file, we create a NAMD job.
line 67 dax.addDependency(Dependency(parent=namdEq_jobOld, child=namdEq_job)) ### The PARENT-CHILD relation is defined here.
...
We see from the dax-generator script it is easy to take the single job script and build the script for N-sequential jobs. In fact, we could take any dax-generator script and modify it for new workflow. This is because the abstraction layer provided by pegasus is a great strength in re-using the workflow or in modifying a workflow to fit closely related computational tasks. If we do not change the scratch and output directories, there is no need to change the sites-generator.script. Next, we will work on the submit.bash file.
Job submission and status
Since the dax-generator is "dax-generator-namdEq-sequential.py", we should have the file specified in the submit.bash script to generate the dax.xml file. Edit submit.bash as follows
...
line 9 ./dax-generator-namdEq-sequential.py ### The dax-generator script is executed and it creates dax.xml file.
...
As mentioned before, we can submit and check the status of the job as follows
### To submit the job
$ ./submit.bash
### To check the status
$ pegasus_status
### or you can also check with the condor_q command
$ condor_q username ### username is your login ID
Excercise-3
Go to the directory "tutorial-pegasus-namd/Exercises/NSeqEx3". All the input files related to run 1,000 sequential NAMD jobs are in the file "inputsN1000.tar.gz". The input files were already generated to save time. Uncompress the files by running the command "tar -xvzf inputsN1000.tar.gz" that will create a directory "inputs" containing all the NAMD input files. The dax-generator python script needs to know about these 1,000 input files, include this information in the dax-generator.
M-Parallel, N-Sequential jobs
We consider the case of running large molecular dynamics simulations of a protein in multiple temperature. The solution is to generate M parallel simulations corresponding to different temperatures. Each one of the parallel simulations contains N-sequential jobs. We can take the N-sequential workflow as template and modify the workflow to fit the M-parallel, N-sequential simulations.
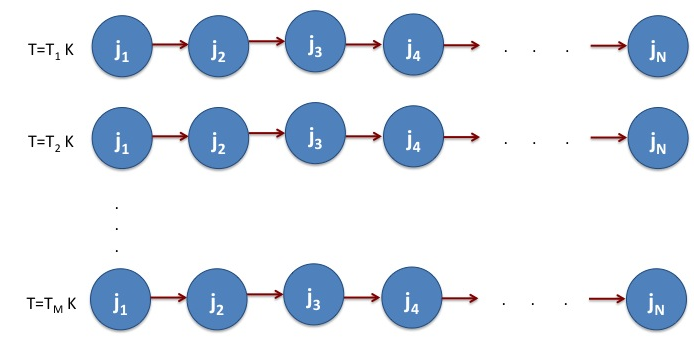
Figure 3. The NAMD simulation is performed for M temperatures - T1, T2, ….TM. For each temperature, there are N-sequential jobs.
Generating M*N input files
We use a script to do the task of generating M*N input files for M-parallel, N-Sequential molecular dynamics simulation.
$ cd tutorial-pegasus-namd/M_times_NSeq ### Directory of required files to run M-parallel, N-sequential MD jobs
$ cd InputGen ### the input template and input generation script are available in this directory
$ ./namd_gen_pegasus_input.bash 12 20 ### This would generate 12 independent MD simulations, each of 20 sequential config fils under the directory inputs
The "namd_gen_pegasus_input.bash" generates M parallel, each of N sequential NAMD inputs. A given parallel job containing N-sequential jobs will be carried out with a particular temperature that was generated from a random process. DAX-generator We have to change our dax.xml via the dax-generator script to account that there are N-sequential jobs. Let us see where is our dax generator file.
$ cd tutorial-pegasus-namd/M_times_NSeq ### Main directory
$ ls dax*.py
dax-generator-namdEq-MtimesNSeq.py ### This is the dax generator file.
Let us analyze the dax-generator script to find what are the primary differences in running M-parallel, N-sequential jobs and running N-sequential jobs.
...
line 8 Mjobs = 16 ### Defines the number of M-parallel jobs (for the current example, this number is 12)
line 9 Njobs = 24 ### Defines the number of N-Sequential jobs (for the current example, this number is 24)
...
line 36 for j in range(1,Mjobs+1): ### The loop for M-parallel jobs
...
The dax-generator for M-parallel, N-sequential jobs is adapted from the dax-generator script to run N-sequential jobs by adding an additional loop. Since the path of scratch and output directories are defined from the relative path of the working directory, there is no need to change the sites-generator.script. Next, we will work on the submit.bash file.
Job submission and status
Since the dax-generator is "dax-generator-namdEq-sequential.py", we should have the file called in the submit script to generate the dax.xml file.
...
line 9 ./dax-generator-namdEq-MtimesNSeq.py ### The dax-generator script is executed and it creates dax.xml file.
...
We submit the jobs and check the status of the job as follows
### To submit the job
$ ./submit.bash
### To check the status
$ pegasus_status
### or you can also check with the condor_q command
$ condor_q username ### username is your login ID
Excercise-4
Go to the directory "tutorial-pegasus-namd/Exercises/MtimesNseqEx4". All the input files related to run 1000 sequential NAMD jobs are in the file "inputsM1000N50.tar.gz". The input files were already generated to save time. Uncompress the files by running the command "tar -xvzf inputsM1000N50.tar.gz" that will create a directory "inputs" containing all the NAMD input files. The input files represent 1000-parallel, each of 50-sequential NAMD jobs. Include this information in the dax-generator.
Keypoints
-
Pegasus requires dax.xml, sites.xml and pegasusrc files. These files contain the information about executable, input and output files and the relation between them while executing the jobs.
-
It is convenient to generate the xml files via scripts. In our example, dax.xml is generated via python script and sites.xml is generated via bash script.
-
To implement a new workflow, edit the existing dax-generator, sites-generator and submit scripts. In the above examples, we modified the workflow for the single NAMD job to implement the workflows of N-sequential and M-parallel, N-sequential jobs.
References
-
OSG QuickStart. Getting started with the Open Science Grid (OSG)
-
HTCondor Manual. Manual for the High Throughput Condor software to schedules the jobs on OSG
*For further assistance or questions, please email connect-support@opensciencegrid.org.